
About ALS

Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease or motor neuron disease, is a rare, chronic, progressive neurodegenerative disease that affects people worldwide, with approximately 20,000 in the United States alone. The average life span from diagnosis is 2 to 5 years, and the majority of ALS cases (~90%) are without a family history of the disease. However, ~10% of ALS cases are caused by inherited genetic mutations (often called familial ALS).
Several genes have been discovered that are causative for ALS, including the gene-encoding sigma-1 receptor (S1R). Mutations resulting in complete loss of function of the S1R are associated with severe, juvenile ALS, while mutations resulting in partial, incomplete function of the S1R are associated with adult onset ALS. This suggests that the S1R is important for the normal function of motor neurons.
{kind=link}

In patients with ALS, motor neurons in the brain and spinal cord that convey messages to the muscles degenerate, affecting the brain’s ability to communicate with muscles. This leads to muscle wasting and progressive paralysis. Patients rapidly lose their ability to walk, speak, eat, and breathe and become fully dependent on their caretakers.
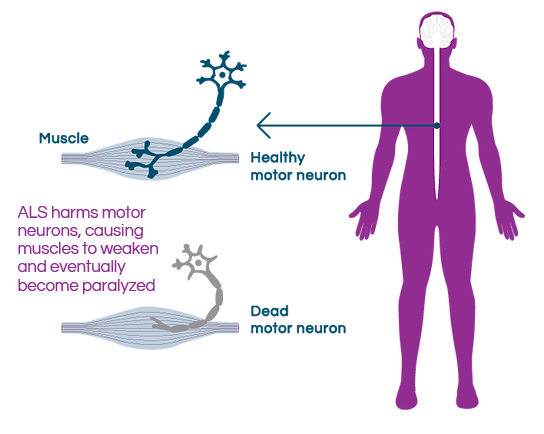
ALS is a devastating disease
for patients, their families, and
caretakers.
ALS is a devastating disease
for patients, their families, and
caretakers.
Pridopidine was assessed in the HEALEY ALS platform study for the treatment of ALS.
While pridopidine did not meet the primary endpoint of change from baseline to week 24 in the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R), there were consistent, positive findings observed among participants receiving pridopidine across several pre-specified secondary and exploratory measures. These results provide valuable insights that will inform future clinical development for pridopidine in ALS.
ACT for ALS Expanded Access to Pridopidine Program
The NIH NINDS-sponsored ACT for ALS Expanded Access to Pridopidine Protocol (EAP), led by the Mass General’s Sean M. Healey & AMG Center for ALS, is available at treating sites across the U.S. for up to 200 ALS individuals who are not eligible for other clinical trials. Those accepted into the EAP may be treated with pridopidine for up to two years. Click here to find more information on eligibility criteria and your nearest site.
We believe pridopidine is a promising drug candidate for the treatment of ALS for several reasons:

Recent data show that some individuals with decreased or loss of function in the sigma-1 receptor develop ALS.

Pridopidine activates sigma-1 receptor and improves motor neuron survival and muscle function in animal models of ALS.

Supportive clinical data from the Phase 2 study of pridopidine in ALS.

Pridopidine is an oral investigational drug with an established safety profile.
These findings highlight the therapeutic potential of pridopidine for treating ALS. In addition, Prilenia holds orphan drug designation for pridopidine in ALS in both the United States and EU.
Pridopidine is an investigational product and has not been approved by the US Food & Drug Administration or other ex-US regulatory agencies. Its safety and efficacy have not yet been determined.